|
Wstęp
Do dystrofii śródbłonka rogówki, określanych również jako tylne dystrofie
rogówki, zalicza się: dystrofię Fuchsa (Fuchs endothelial corneal dystrophy –
FECD), tylną polimorficzną dystrofię rogówki (posterior polymorphus corneal
dystrophy – PPCD) i wrodzone dziedziczne dystrofie śródbłonka (congenital
hereditary endothelial dystrophy – CHED). Śródbłonek rogówki tworzą komórki
pochodzące z grzebienia nerwowego i uważa się, że ta grupa chorób rogówki
reprezentuje defekt końcowego różnicowania komórek grzebienia nerwowego (1).
Dystrofie te powstają więc na skutek pierwotnej dysfunkcji śródbłonka rogówki,
a ich wspólnymi cechami są metaplazja komórek śródbłonka i tworzenie się
nieprawidłowej błony Descemeta (2,3).
Podobieństwa te sprawiły, że ostatnio poszukuje się również wspólnego podłoża
patogenetycznego dla tej grupy chorób. Identyfikacja genu, którego mutacje
wykrywa się w jednej z tych dystrofii, inspiruje badaczy do poszukiwania
mutacji w tym samym genie w pozostałych dystrofiach z tej grupy. Dzieje się tak
zwłaszcza w przypadku dystrofii Fuchsa i PPCD, ale również PPCD i CHED1.
Próby te mogą się wydawać dość zaskakujące ze względu na różnice zarówno w
przebiegu klinicznym, jak i morfologii zmian obserwowanych w tych dystrofiach.
Tutaj warto jednak wspomnieć o genie TGFBI (transforming growth factor
beta-induced), którego mutacje są wykrywane w różnych dystrofiach istoty
właściwej rogówki, dystrofiach warstwy Bowmana, ale również w części przypadków
dystrofii błony podstawnej nabłonka (4). Przykład genu TGFBI potwierdza, że
nawet jednostki chorobowe różniące się od siebie przebiegiem klinicznym,
lokalizacją i morfologią zmian mogą być powodowane przez mutacje w tym samym
genie.
Czy w związku z tym dystrofie śródbłonka rogówki mogą być również konsekwencją
oddziaływania różnych wariantów allelicznych jednego genu? Jakie geny odgrywają
istotną rolę w ich patogenezie? Odpowiedzi na te pytania szukaliśmy, analizując
aktualny stan wiedzy na temat podłoża genetycznego dystrofii rogówki.
Dystrofia Fuchsa (FECD)
Dystrofie Fuchsa występują z różną częstością w różnych częściach świata. W
Stanach Zjednoczonych i innych krajach rozwiniętych jest to najczęstsza
pierwotna choroba rogówki, która dotyka ok. 4% populacji. U kobiet występuje
prawie 3-4 razy częściej niż u mężczyzn, jej przebieg u kobiet jest znacznie
cięższy. Natomiast w Arabii Saudyjskiej, u Chińczyków z Singapuru i w Japonii
ta postać dystrofii jest niezwykle rzadka (5). Zdecydowana większość pacjentów
z dystrofią Fuchsa ma objawy choroby najwcześniej około 5. dekady życia i
negatywny wywiad rodzinny. Jednak u części chorych wywiad rodzinny jest
obciążony, wskazuje na autosomalny dominujący typ dziedziczenia, a objawy
choroby mogą się pojawiać już w pierwszej dekadzie życia lub dopiero w czwartej
i późniejszych (4). Dystrofia Fuchsa ma charakter postępujący i cechuje się
obecnością zmian określanych jako „cornea guttata”. Są to załamujące światło „wyrośla”
błony Descemeta, która jest warstwą rogówki bogatą w kolagen wydzielany przez
śródbłonek.
Gen COL8A2, zlokalizowany na krótkim ramieniu chromosomu 1. (1p34.3-p32.3),
koduje łańcuch alpha 2 kolagenu 8., będący składnikiem macierzy pozakomórkowej.
Białko to pełni rolę strukturalną, ale przypisuje mu się również rolę w
procesie różnicowania komórek (6,7). W dystrofii Fuchsa skupiska tego właśnie
kolagenu wykrywano w tylnej części błony Descemeta, tworzącej błonę podstawną
komórek śródbłonka. Sugerowało to jego rolę w powstawaniu typowych dla
dystrofii Fuchsa zmian typu „guttae” (8).
Zwłaszcza w przypadkach rodzinnych dystrofii Fuchsa o wczesnym początku („early-onset”)
identyfikowano mutacje w genie COL8A2. Były to mutacje typu zmiany sensu:
zamiana leucyny na tryptofan (Leu450W) oraz zamiana glutaminy na lizynę (Q455K)
(9,10). Pacjenci z tymi mutacjami prezentowali również charakterystyczny
fenotyp. Zmiany typu „guttae” były u nich nieduże, nieznacznie uniesione i
zlokalizowane w pobliżu środka komórek śródbłonka (ryc. 1). Obszarom o dużym
zagęszczeniu zmian towarzyszyły rozległe obszary pozbawione „guttae”. W
przeciwieństwie do tego „guttae” u pacjentów z późną postacią dystrofii Fuchsa
były wyżej uniesione i lokalizowały się w pobliżu połączeń międzykomórkowych
przy powierzchni bocznopodstawnej komórek śródbłonka (9).
Ponadto u pojedynczych osób z dystofią Fuchsa o późnym początku wykryto trzy
inne mutacje typu zmiany sensu w COL8A2: zamianę argininy na glutaminę (R155Q i
R304Q) oraz zamianę argininy na histydynę (R434H) (10). Kolejne badania
pokazały jednak, że mutacje w COL8A2
dotyczą tylko niedużej grupy pacjentów, a niektóre zmiany w tym genie, tj.
R155Q, wykrywano również u osób zdrowych, poddając w wątpliwość jej patogenność
(11,12). Mutacje w COL8A2 zatem są odpowiedzialne z rozwój dystrofii Fuchsa o
wczesnym początku, ale takiego powiązania nie znaleziono dla znacznie częstszej
postaci dystrofii Fuchsa, dystrofii o późnym początku.
U pacjentów z dystrofią Fuchsa poszukiwano również mutacji w genie ZEB1 (TCF8)
związanym patogenetycznie z inną dystrofią śródbłonka rogówki – dystrofią
polimorficzną tylną. Tylko u jednego pacjenta spośród 74 osób znaleziono
heterozygotyczną nową zmianę N696S w genie ZEB1 (TCF8) (13). Wyniki tych badań
sugerowały, że ZEB1 (TCF8) nie odgrywa istotnej roli w patogenezie dystrofii
Fuchsa. Jednak ostatnio ukazała się publikacja potwierdzająca związek dystrofii
Fuchsa z mutacjami w genie ZEB1 (TCF8). Są to mutacje typu zmiany sensu, które
prowadzą do utraty funkcji kodowanego przez ten gen czynnika transkrypcyjnego.
Obecność mutacji Q840P w genie ZEB1 (TCF8) zmieniającej glutaminę w prolinę
jest wystarczająca do rozwoju dystrofii Fuchsa (14).
Podobnie u pacjentów z dystrofią Fuchsa szukano mutacji w genie SLC4A11,
związanym z rozwojem dziedzicznej dystrofii śródbłonka (CHED2). Vithana i wsp.
(15) badali pacjentów pochodzących z Singapuru, Hongkongu, Indii i tylko u
czterech osób spośród 89 pacjentów z tą dystrofią, a więc u niespełna 5%,
znaleziono mutacje w genie SLC4A11. Trzy z nich to heterozygotyczne mutacje
typu zmiany sensu (E399K, G709E i T754M), a jedna to delecja (c.99-100delTC).
Sprawdziliśmy w bazie mutacji genetycznych człowieka (www.hgmd.cf.ac.uk/ac/index.php),
że są to inne zmiany niż te, dotychczas opisywane u pacjentów z CHED2, chorobie
o dziedziczeniu autosomalnym recesywnym. W przeciwnym razie należałoby się
spodziewać, że wszyscy rodzice dzieci z CHED2 będą chorowali na dystrofię
Fuchsa.
Intensywne poszukiwanie podłoża genetycznego dystrofii Fuchsa doprowadziło
również do identyfikacji kilku podejrzanych obszarów chromosomowych (locus,
l.mnoga loci), w obrębie których obecnie intensywnie poszukuje się genów
odpowiedzialnych za rozwój tej dystrofii. Są to loci na chromosomie 13.
(13pTel-13q12.13) (16), a także długim ramieniu chromosomów: 15. (12) i 18.
(18q21.2-q21.32) (13), a ostatnio również chromosomu 5. (5q33.1-q35.2) (17) i
krótkiego ramienia chromosomu 9. (14). Zauważono, że obecność określonego
haplotypu w obszarze 9p w połączeniu z obecnością wcześniej wspomnianej mutacji
Q840P w genie ZEB1 (TCF8) wiąże się z rozwojem ciężkiej postaci dystrofii
Fuchsa (14). Biorąc pod uwagę powyższe, należy sądzić, że dystrofia Fuchsa o
późnym początku jest chorobą heterogenną genetycznie, powodowaną przez
interakcje produktów różnych genów (5).
Dystrofia polimorficzna tylna (PPCD)
PPCD (czasem określana również skrótem PPMD) charakteryzuje się obecnością
pogrubiałej błony Descemeta i występowaniem w miejscu komórek śródbłonka
komórek o właściwościach nabłonka wielowarstwowego z mikrokosmkami i
połączeniami desmosomalnymi (5). Dystrofia ta dziedziczy się w sposób
autosomalny dominujący i ma bardzo zmienną ekspresję obrazu klinicznego.
Spektrum zmian fenotypowych w PPCD obejmuje bezobjawowe zmiany śródbłonka
rogówki, pogrubienie błony Descementa, występujące u większości pacjentów, a w
przypadkach o ciężkim przebiegu może dojść do obrzęku rogówki (ryc 2). U około
15% pacjentów z PPCD może się rozwinąć jaskra (4).
Dotychczas zidentyfikowano trzy loci chromosomowe związane z rozwojem tej
choroby. PPCD1 obejmuje obszar okołocentromerowy chromosomu 20 (20p11.2-q11.2)
(18-20), PPCD2 – obszar na krótkim ramieniu chromosomu 1. (1p34.3-p32.3), a
PPCD3 – obszar na krótkim ramieniu chromosomu 10. (10p11.2) (21).
W obrębie locus PPCD1 znajduje się między innymi gen VSX1 (visual system
homeobox gene 1) kodujący czynnik transkrypcyjny biorący udział w regulacji
różnicowania tkanek narządu wzroku. Dotychczas tylko w dwóch rodzinach z PPCD
zidentyfikowano mutacje w genie VSX1 (22,23) i obecnie ich znaczenie w rozwoju
tej choroby uznaje się raczej za marginalne (5,19,24).
Niezwykle interesujący jest natomiast fakt, że w obrębie locus PPCD1
poszukiwany jest również gen odpowiedzialny za rozwój CHED1. Obie jednostki
chorobowe wykazują znaczne podobieństwa kliniczne i histologiczne (5,25), a
krewni osób z PPCD mogą mieć CHED1 (26,27). Biorąc pod uwagę różnorodność loci
chromosomowych dla PPCD, należy przypuszczać, że w różnych rodzinach do rozwoju
tej choroby mogą prowadzić inne geny. Wydaje się jednak prawdopodobne, że
poszukiwany gen na chromosomie 20. mógłby być wspólny dla PPCD i CHED1.
Niektórzy badacze sugerują nawet, że CHED1 może być odmianą PPCD o wczesnym
początku i ciężkim przebiegu, a nie oddzielną jednostką chorobową (24). Na
razie jednak, pomimo intensywnych poszukiwań, nie znaleziono żadnej mutacji w
regionach kodujących ponad 35 różnych genów znajdujących się w obszarze PPCD1
(24).
W locus PPCD2 znajduje się gen COL8A2, którego mutacje, jak pisaliśmy powyżej,
prowadzą do rozwoju dystrofii Fuchsa o wczesnym początku. Biswas i wsp. (10)
publikując pierwsze doniesienie o związku COL8A2 z dystrofią Fuchsa, opisali
również mutację Q455K w tym genie w rodzinie z PPCD. Poza tym jednym
doniesieniem brakuje innych dowodów takiej zależności i rola mutacji COL8A2 w
patogenezie PPCD pozostaje na razie niejasna (28).
W przeciwieństwie do wątpliwości dotyczących VSX1 i COL8A2 coraz bardziej
przekonująca jest rola ZEB1 (TCF8) w rozwoju PPCD. TCF8 (transcription factor
8) został zidentyfikowany w locus PPCD3 na chromosomie 10.
(29). Zgodnie z obowiązującą nomenklaturą przyjętą nazwą tego genu jest symbol
ZEB1 (zinc-finger E-box binding homeobox 1) (http://www.genenames.org/index.html),
ale w wielu publikacjach nadal często spotyka się symbol TCF8. U pacjentów z
PPCD w genie ZEB1 (TCF8) identyfikuje się mutacje typu „nonsense” wprowadzające
kodon stop i kończące ramkę odczytu, bądź delecje lub duplikacje, które
przesuwają ramkę odczytu i zmieniają sekwencję aminokwasową kodowanego białka
(29-31).
Gen ZEB1 (TCF8) koduje czynnik transkrypcyjny – białko wiążące się z DNA i
regulujące ekspresję genów. Jednym z genów docelowych dla białka ZEB1 (TCF8)
jest COL3A4, kodujący łańcuch alpha 3 kolagenu 4. (29). Z kolei mutacje w genie
COL3A4 prowadzą do powstania zespołu Alporta, którego jedną z cech, obserwowaną
u części pacjentów, jest PPCD (29).
Zwrócono uwagę, że u pacjentów z mutacjami ZEB1 (TCF8), częściej niż w
populacji ogólnej, występują przepukliny (pachwinowe, brzuszne i pępkowe),
wodniaki oraz nieprawidłowości układu kostnego, tj. dodatkowe kręgi, wyrośla
kostne na kręgach kręgosłupa, guzki rzepki, guzki na kościach dłoni i stóp,
związane z przykurczami Dupuytrena, oraz choroba Osgooda-Schlattera (aseptyczne
zapalenie guzowatości kości piszczelowej) (29,32).
Zależnie od badanej grupy mutacje w ZEB1 (TCF8) wykrywa się u 10-50% pacjentów
z PPCD (29-31). Mutacje w ZEB1 (TCF8) znaleziono u około połowy badanych
pacjentów w Stanach Zjednoczonych Ameryki i Australii, u członków wszystkich
badanych rodzin w Wielkiej Brytanii, ale nie znaleziono ich u żadnego z
pacjentów pochodzących z Czech (29,30), co wskazuje na udział jeszcze innych,
niepoznanych dotychczas, genów w patogenezie PPCD.
Wrodzone dziedziczne dystrofie śródbłonka (CHED)
U pacjentów z CHED rogówka jest matowa i znacznie pogrubiała (nawet 2-3 razy w
porównaniu z prawidłową) na skutek obrzęku istoty właściwej. Chorobę rozpoznaje
się w dwóch pierwszych latach życia. Do objawów CHED należą łzawienie i
światłowstręt, a u pacjentów z CHED2 – dodatkowo oczopląs. Ponadto CHED1 ma
przebieg postępujący, podczas gdy przebieg CHED2 jest stabilny.
Histopatologicznie CHED1 i 2 są podobne, jednak w CHED1 tylna warstwa błony
Descemeta jest pogrubiała na skutek odkładanego tam kolagenu, podczas gdy w
CHED2 jest to spowodowane produkcją homogennej masy przez nieprawidłowe komórki
śródbłonka. Podobnie jak w PPCD u pacjentów z CHED komórki śródbłonka wykazują
pewne cechy komórek nabłonkowych, ale znacznie rzadziej tworzą nabłonek
wielowarstwowy (4,5).
CHED1 dziedziczy się autosomalnie dominująco, ale genu odpowiedzialnego za
wystąpienie tej choroby dotychczas nie znaleziono. Tak jak pisaliśmy powyżej,
na podstawie analizy sprzężeń zidentyfikowano locus dla CHED1 w regionie
okołocentromerowym chromosomu 20., które obejmuje również część locus dla PPCD
(24,33).
CHED2 dziedziczy się autosomalnie recesywnie, występuje częściej i ma przebieg
cięższy niż CHED1 (4). Związany z rozwojem CHED2 gen SLC4A11 (solute carrier
family 4,
member 11) został odkryty w roku 2006 przez Vithannę i wsp.(34). Ten gen,
zlokalizowany na krótkim ramieniu chromosomu 20. (20p13-p12) koduje białko
transbłonowe uczestniczące w transporcie jonów boranowych, co jest niezbędne
dla komórkowej homeostazy boru wykorzystywanego przez komórki do wzrostu i
proliferacji (35).
Niedawno została opisana również dystrofia rogówki sprzężona z chromosomem X (XECD,
dziedziczenie dominujące), której objawy są widoczne przy urodzeniu, a przebieg
choroby jest cięższy u chłopców. Locus dla XECD zidentyfikowano na długim
ramieniu chromosomu X (Xq25), ale gen odpowiedzialny za wystąpienie tej choroby
nie został na razie znaleziony (36).
Podsumowanie
W patogenezie dystrofii śródbłonka rogówki niezwykle ciekawa wydaje się rola
genu ZEB1 (TCF8). Mutacje tego genu są wykrywane w PPCD, ale według najnowszych
doniesień ma on również znaczenie w rozwoju dystrofii Fuchsa (14). Czy zatem
dystrofia Fuchsa (przynajmniej część przypadków) i PPCD są różnymi obrazami
morfologicznymi defektów tego samego genu? Na razie, naszym zdaniem, jest zbyt
wcześnie na udzielenie jednoznacznie twierdzącej odpowiedzi. W tabeli I
przedstawiliśmy dotychczas poznane loci chromosomowe i geny związane z rozwojem
dystrofii śródbłonka rogówki.
Piśmiennictwo:
1. Bahn CF, Falls HF, Varley GA, Meyer RF, Edelhauser HF,
Bourne WM: Classification of corneal endothelial disorders based on neural
crest origin. Ophthalmology 1984, 91(6), 558-563.
2. Levy SG, Moss J, Sawada H, Dopping-Hepenstal PJ, McCartney AC: The
composition of wide-spaced collagen in normal and diseased Descemet’s membrane.
Curr Eye Res 1996, 15(1), 45-52.
3. McCartney AC, Kirkness CM: Comparison between posterior polymorphous
dystrophy and congenital hereditary endothelial dystrophy of the cornea. Eye (Lond)
1988, 2 (Pt 1), 63-70.
4. Weiss JS, Moller HU, Lisch W, Kinoshita S, Aldave AJ, Belin MW et al.: The
IC3D classification of the corneal dystrophies. Cornea 2008, 27 Suppl 2, S1-83.
|
|
5. Klintworth GK: Corneal dystrophies. Orphanet J Rare Dis
2009, 4, 7.
6. Levy SG, Moss J, Sawada H, Dopping-Hepenstal PJ, McCartney AC: The
composition of wide-spaced collagen in normal and diseased Descemet’s membrane.
Curr Eye Res 1996, 15(1), 45-52.
7. Sage H, Iruela-Arispe ML: Type VIII collagen in murine development.
Association with capillary formation in vitro. Ann N Y Acad Sci 1990, 580,
17-31.
8. Kenney MC, Labermeier U, Hinds D, Waring GO III: Characterization of the
Descemet’s membrane/posterior collagenous layer isolated from Fuchs’
endothelial dystrophy corneas. Exp Eye Res 1984, 39(3), 267-277.
9. Gottsch JD, Sundin OH, Liu SH, Jun AS, Broman KW, Stark WJ et al.:
Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype
of fuchs corneal dystrophy. Invest Ophthalmol Vis Sci 2005, 46(6), 1934-1939.
10. Biswas S, Munier FL, Yardley J, Hart-Holden N, Perveen R, Cousin P et al.:
Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII
collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet 2001,
10(21), 2415-2423.
11. Kobayashi A, Fujiki K, Murakami A, Kato T, Chen LZ, Onoe H et al.: Analysis
of COL8A2 gene mutation in Japanese patients with Fuchs’ endothelial dystrophy
and posterior polymorphous dystrophy. Jpn J Ophthalmol 2004, 48(3), 195-198.
12. Afshari NA, Li YJ, Pericak-Vance MA, Gregory S, Klintworth GK: Genome-wide
linkage scan in fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci
2009, 50(3), 1093-1097.
13. Sundin OH, Broman KW, Chang HH, Vito EC, Stark WJ,
Gottsch JD: A common locus for late-onset Fuchs corneal dystrophy maps to
18q21.2-q21.32. Invest Ophthalmol Vis Sci 2006, 47(9), 3919-3926.
14. Riazuddin SA, Zaghloul NA, Al Saif A, Davey L, Diplas BH,
Meadows DN et al.: Missense Mutations in TCF8 Cause Late-Onset Fuchs Corneal
Dystrophy and Interact with FCD4 on Chromosome 9p. Am J Hum Genet 2009.
15. Vithana EN, Morgan PE, Ramprasad V, Tan DT, Yong VH,
Venkataraman D et al.: SLC4A11 mutations in Fuchs endothelial corneal dystrophy.
Hum Mol Genet 2008, 17(5), 656-666.
16. Sundin OH, Jun AS, Broman KW, Liu SH, Sheehan SE, Vito EC et al.: Linkage
of late-onset Fuchs corneal dystrophy to a novel locus at 13pTel-13q12.13.
Invest Ophthalmol Vis Sci 2006, 47(1), 140-145.
17. Riazuddin SA, Eghrari AO, Al Saif A, Davey L, Meadows DN, Katsanis N et
al.: Linkage of a mild late-onset phenotype of Fuchs corneal dystrophy to a
novel locus at 5q33.1-q35.2. Invest Ophthalmol Vis Sci 2009, 50(12), 5667-5671.
18. Yellore VS, Papp JC, Sobel E, Khan MA, Rayner SA, Farber DB et al.:
Replication and refinement of linkage of posterior polymorphous corneal
dystrophy to the posterior polymorphous corneal dystrophy 1 locus on chromosome
20. Genet Med 2007, 9(4), 228-234.
19. Gwilliam R, Liskova P, Filipec M, Kmoch S, Jirsova K, Huckle EJ et al.:
Posterior polymorphous corneal dystrophy in Czech families maps to chromosome
20 and excludes the VSX1 gene. Invest Ophthalmol Vis Sci 2005, 46(12),
4480-4484.
20. Heon E, Mathers WD, Alward WL, Weisenthal RW, Sunden SL, Fishbaugh JA et
al.: Linkage of posterior polymorphous corneal dystrophy to 20q11. Hum Mol
Genet 1995, 4(3), 485-488.
21. Shimizu S, Krafchak C, Fuse N, Epstein MP, Schteingart MT, Sugar A et al.:
A locus for posterior polymorphous corneal dystrophy (PPCD3) maps to chromosome
10. Am J Med Genet A 2004, 130A(4), 372-377.
22. Heon E, Greenberg A, Kopp KK, Rootman D, Vincent AL,
Billingsley G et al.: VSX1: a gene for posterior polymorphous dystrophy and
keratoconus. Hum Mol Genet 2002, 11(9), 1029-1036.
23. Valleix S, Nedelec B, Rigaudiere F, Dighiero P, Pouliquen Y, Renard G et
al.: H244R VSX1 is associated with selective cone ON bipolar cell dysfunction
and macular degeneration in a PPCD family. Invest Ophthalmol Vis Sci 2006,
47(1), 48-54.
24. Aldave AJ, Yellore VS, Vo RC, Kamal KM, Rayner SA, Plaisier CL et al.:
Exclusion of positional candidate gene coding region mutations in the common
posterior polymorphous corneal dystrophy 1 candidate gene interval. Cornea
2009, 28(7), 801-807.
25. McCartney AC, Kirkness CM: Comparison between posterior polymorphous
dystrophy and congenital hereditary endothelial dystrophy of the cornea. Eye (Lond)
1988, 2 ( Pt 1), 63-70.
26. Levenson JE, Chandler JW, Kaufman HE: Affected asymptomatic relatives in
congenital hereditary endothelial dystrophy. Am J Ophthalmol 1973, 76(6),
967-971.
27. Kanai A, Waltman S, Polack FM, Kaufman HE: Electron microscopic study of
hereditary corneal edema. Invest Ophthalmol 1971, 10(2), 89-99.
28. Yellore VS, Rayner SA, Emmert-Buck L, Tabin GC, Raber I, Hannush SB et al.:
No pathogenic mutations identified in the COL8A2 gene or four positional
candidate genes in patients with posterior polymorphous corneal dystrophy.
Invest Ophthalmol Vis Sci 2005, 46(5), 1599-1603.
29. Krafchak CM, Pawar H, Moroi SE, Sugar A, Lichter PR, Mackey DA et al.:
Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic
expression of COL4A3 by corneal endothelial cells. Am J Hum Genet 2005, 77(5),
694-708.
30. Liskova P, Tuft SJ, Gwilliam R, Ebenezer ND, Jirsova K,
Prescott Q et al.: Novel mutations in the ZEB1 gene identified in Czech and
British patients with posterior polymorphous corneal dystrophy. Hum Mutat 2007,
28(6), 638.
31. Vincent AL, Niederer RL, Richards A, Karolyi B, Patel DV, McGhee CN:
Phenotypic characterisation and ZEB1 mutational analysis in posterior
polymorphous corneal dystrophy in a New Zealand population. Mol Vis 2009, 15,
2544-2553.
32. Aldave AJ, Yellore VS, Yu F, Bourla N, Sonmez B, Salem AK et al.: Posterior
polymorphous corneal dystrophy is associated with TCF8 gene mutations and
abdominal hernia. Am J Med Genet A 2007, 143A(21), 2549- -2556.
33. Toma NM, Ebenezer ND, Inglehearn CF, Plant C, Ficker LA, Bhattacharya SS:
Linkage of congenital hereditary endothelial dystrophy to chromosome 20. Hum
Mol Genet 1995, 4(12), 2395-2398.
34. Vithana EN, Morgan P, Sundaresan P, Ebenezer ND, Tan DT, Mohamed MD et al.:
Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital
hereditary endothelial dystrophy (CHED2). Nat Genet 2006, 38(7), 755-757.
35. Park M, Li Q, Shcheynikov N, Zeng W, Muallem S: NaBC1 is a ubiquitous
electrogenic Na+ -coupled borate transporter essential for cellular boron
homeostasis and cell growth and proliferation. Mol Cell 2004, 16(3), 331-341.
36. Schmid E, Lisch W, Philipp W, Lechner S, Gottinger W, Schlotzer-Schrehardt
U et al.: A new, X-linked endothelial corneal dystrophy. Am J Ophthalmol 2006,
141(3), 478-487.



Ryc. 1. Nasilone zmiany „guttae” w śródbłonku 40-letniej
pacjentki – a. oświetlenie bezpośrednie, b. retroiluminacja, c. mikroskopia
konfokalna.
Fig. 1. Escalated lesions of „guttae” type in the endothelium of 40-years old
female –
a. direct illumination, b. retroillumination, c. confocal microscopy.

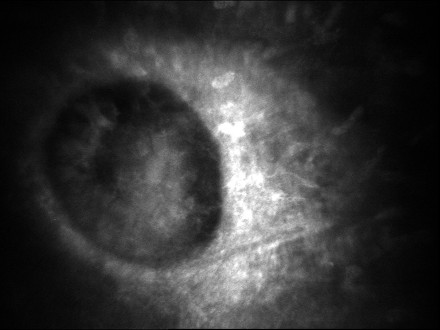

Ryc. 2. Zmiany charakterystyczne dla dystrofii polimorficznej
tylnej w śródbłonku i błonie Descemeta 10-letniej pacjentki – a. oświetlenie
bezpośrednie, b., c. mikroskopia konfokalna.
Fig. 2. Lesions characteristic for Posterior Polymorphous Dystrophy within the
endothelium of 10-years old female patient – a. direct illumination, b., c.
confocal microscopy.
|
|








